


量子化學:分子軌道中的開殼與閉殼
在量子化學計算中,“開殼(open-shell)”與“閉殼(closed-shell)”是兩個經常出現卻容易混淆的概念。它們不僅反映了體系的電子構型,更決定了自旋狀態、反應活性、磁性乃至計算結果的可靠性。尤其在 Gaussian 計算中,正確區分體系是開殼還是閉殼,不僅影響能量的準確性,也關系到計算是否能正常收斂。本文將從概念出發,結合 Gaussian 的實際應用,帶你系統理解這兩種的“根本差異”。

一、閉殼體系
閉殼體系(Closed-shell system)指所有軌道都被成對電子填滿,體系總自旋為零,通常對應單重態(Singlet)。
這類體系在能量上往往較低,化學性質穩定,如 H?、CH?、CO? 等分子都屬于典型的閉殼體系。在 Gaussian 計算中,閉殼體系默認采用限制性(Restricted)方法,即α、β電子占據相同軌道。例如常用的 RHF 或 R-B3LYP,計算速度快、收斂穩定、結果可靠。判斷一個體系是否為閉殼,可從以下幾點入手:
自旋多重度(Multiplicity) = 1;
輸出文件中
≈ 0; 自旋密度圖上無明顯未配對電子。
這類體系常用于描述穩定的中性分子、離子對、以及多數過渡態優化的初始結構。
二、開殼體系
與閉殼體系不同,開殼體系(Open-shell system)中存在一個或多個未配對電子,因此體系總自旋不為零。它們通常表現為雙重態(doublet)、三重態(triplet)或更高自旋態,常見于自由基、金屬中心以及激發態分子。開殼體系的存在,是許多化學反應——尤其是氧化還原、光化學、自由基與過渡金屬催化反應——的關鍵。在 Gaussian 中,開殼體系的計算通常采用非限制性(Unrestricted)方法,即允許 α、β 電子分布不同。例如:UB3LYP、UHF、UM06-2X 等方法都可用于開殼體系。在輸出結果中,判斷開殼體系是否合理計算的關鍵是
雙重態理論值應為 0.75;
三重態為 2.00;
若顯著偏大(如雙重態算得 >1.0),說明出現自旋污染(spin contamination),需要重新檢查計算設置。開殼體系的常見問題與修正
1、自旋污染(Spin contamination)
開殼計算中最常見的問題。若
2、自旋態競爭
某些體系可能存在多個相近能量的自旋態,例如雙重態與四重態同時穩定。此時應分別計算不同自旋態能量,比較后確定真實基態。
3、閉殼假設錯誤
若強行用閉殼方法計算自由基體系,可能導致收斂失敗或得到虛假的低能結果。例如·OH、NO?、ClO 都必須使用開殼方法。
4、激發態與光化學體系
激發態分子往往由閉殼轉為開殼,需要顯式指定自旋多重度,并考慮激發態能量差。
三、開殼與閉殼的判斷
判斷一個體系究竟屬于開殼還是閉殼,是電子結構分析中最容易被誤解的部分。人們常以“偶數電子=閉殼、奇數電子=開殼”作為經驗判斷,但這只在簡單分子中成立。
O? 分子為三重態開殼體系;
NO、ClO 等自由基雖電子數奇數,但可呈穩定雙重態;
某些金屬配合物(如高自旋 Fe(III))即使總電子偶數,仍為開殼態。
這說明,即使電子總數為偶數,只要存在軌道簡并、能級接近或未完全配對,就可能形成開殼結構。
1、 軌道占據決定“殼態”
當分子軌道能級依次被電子完全成對占據時,體系為閉殼;
而若最高占據軌道(HOMO)上存在未配對電子,即為開殼。
2、自旋排列是更深層的判據
在多電子體系中,電子填充遵循洪特規則(Hund’s rule):
電子傾向于平行自旋占據簡并軌道,從而產生凈自旋。
因此某些金屬離子(如 Fe3?、Mn2?)即使電子總數為偶數,仍呈現高自旋開殼態。這也是過渡金屬催化中常見的多自旋態現象。
3、 激發態與近簡并軌道
當體系處于激發態或軌道能級極為接近時,也可能由閉殼轉為開殼。典型如乙烯(C?H?)的激發態,其π→π*激發導致一個電子躍遷至反鍵軌道,形成未配對電子對,即開殼單重態(open-shell singlet)。
4、高級方法的識別方式
在 Gaussian 輸出中,可以通過以下信號判斷體系是否潛在開殼特征:
值偏離 0; 自旋密度圖顯示局域化未配對電子;
分子軌道能級圖(MO diagram)中 HOMO–LUMO 間距極小;
收斂困難或能量震蕩(表明軌道混合與自旋競爭)。
當出現這些跡象,即便體系電子數為偶數,也應嘗試以開殼方法重新計算。
5、模糊邊界:開殼單重態與多組態體系
有些體系介于開殼與閉殼之間,例如:
開殼單重態(Open-shell singlet):兩未配對電子自旋反向配對,但空間分布不同,如羰基羰自由基中間體;
雙重勢能面(Two-state reactivity):金屬催化體系中,高低自旋態能量接近,反應可能跨越不同自旋面進行。
這些情況超出單組態 DFT 的描述范圍,通常需使用多組態方法(如 CASSCF、CASPT2)來捕捉真實電子結構。
四、總結
Gaussian 在輸入文件中通過“電荷(Charge)”與“自旋多重度(Multiplicity)”來確定電子構型:
Charge Multiplicity
例如:
0 1 → 中性分子,單重態(閉殼);
0 2 → 中性分子,自由基雙重態(開殼);
0 3 → 中性分子,三重態(開殼)。
在計算過程中,Gaussian 會根據這個設置選擇相應的波函數類型(R或U),但若手動指定錯誤,可能導致能量異常或無法收斂。
因此建議在每次計算后檢查輸出文件中:
值是否接近理論值; 是否出現 “Convergence failure” 或 “SCF may be unstable”;
若能量曲線劇烈震蕩,可嘗試換用 ROHF 或更合適的泛函。
開殼與閉殼的區別,看似只是“電子是否配對”,實則反映了分子的整個量子特征。在 Gaussian 計算中,
閉殼體系代表穩定、配對、低能量的電子狀態;
開殼體系則是活躍、反應性高、具有自旋特征的體系。
對于研究者而言,判斷體系屬于哪種殼態,是所有計算化學工作的起點。理解殼態之分,就等于掌握了分子電子世界的開關。




 投訴建議
投訴建議
提交

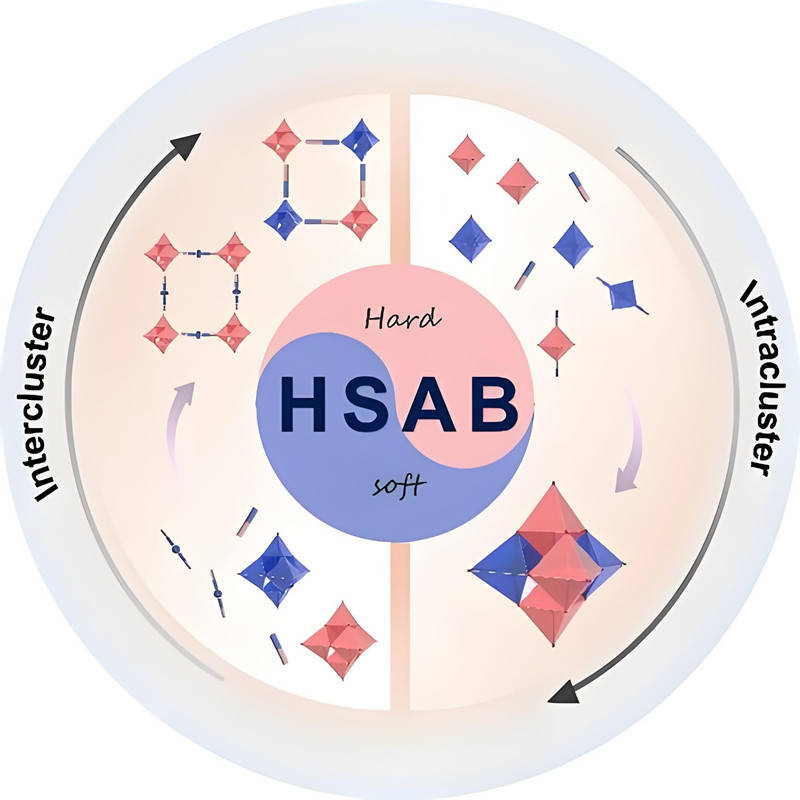
什么是軟硬酸堿理論?
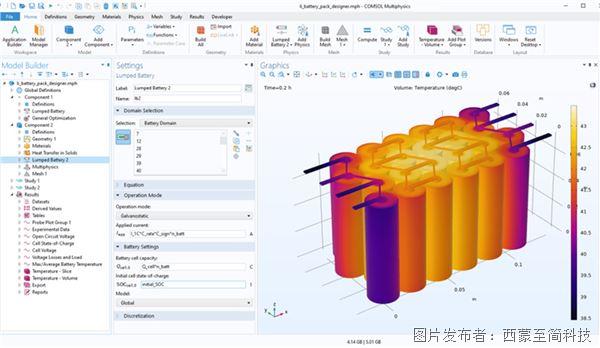
COMSOL有限元仿真,什么是階躍函數
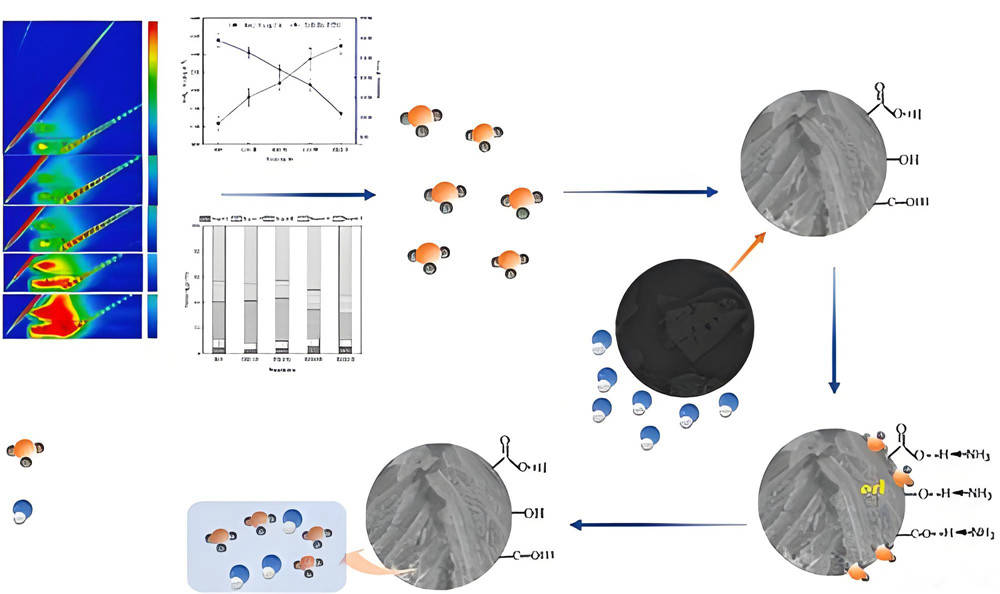
量子化學:什么是吸附機理?
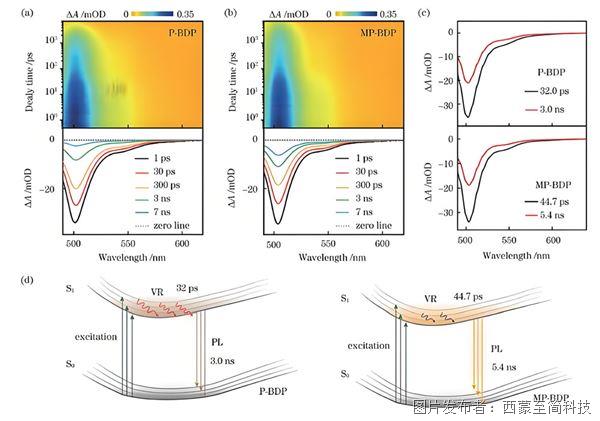
量子化學中的激發態計算的原理與應用
有限元仿真的原理基礎和應用領域!