


量子化學(xué)如何選擇計(jì)算方法與相應(yīng)的基組?
Gaussian是目前應(yīng)用最廣泛的量子化學(xué)計(jì)算軟件之一,既可進(jìn)行半經(jīng)驗(yàn)計(jì)算,也支持從頭算與密度泛函理論等多種計(jì)算類型。該程序幾乎涵蓋了分子體系研究的各個(gè)方面,可用于探索分子能量與幾何結(jié)構(gòu)、過渡態(tài)的構(gòu)型及反應(yīng)勢(shì)壘、化學(xué)鍵性質(zhì)與反應(yīng)能量、分子軌道與電荷密度分布、偶極矩與靜電勢(shì)、多極矩特征、振動(dòng)頻率、紅外與拉曼光譜、核磁共振(NMR)參數(shù)、極化率與超極化率等光學(xué)性質(zhì),以及熱力學(xué)數(shù)據(jù)和反應(yīng)路徑等。
在實(shí)際計(jì)算中,選擇合適的計(jì)算方法與基組是獲得準(zhǔn)確可靠結(jié)果的關(guān)鍵。不同的方法在計(jì)算精度與資源消耗上差異顯著,了解它們的原理和適用范圍有助于根據(jù)研究體系與目標(biāo)做出最優(yōu)決策。以下將對(duì)Gaussian中常用的幾類計(jì)算方法進(jìn)行簡(jiǎn)要介紹與選擇建議。
一、計(jì)算方法的選擇
半經(jīng)驗(yàn)方法(AM1、PM3、CNDO、INDO、MINDO)
通過引入實(shí)驗(yàn)參數(shù)簡(jiǎn)化計(jì)算,速度快但精度有限。
適合:
大體系有機(jī)分子的幾何優(yōu)化;
初步構(gòu)型篩選與趨勢(shì)判斷。
不適用于:
含金屬或強(qiáng)電子相關(guān)體系;
高精度能量計(jì)算。
?建議:僅在體系龐大、計(jì)算資源有限時(shí)用于初步優(yōu)化。
2. 從頭算(Ab initio)方法——Hartree-Fock (HF) 系列
HF方法完全基于量子力學(xué)原理,不含經(jīng)驗(yàn)參數(shù)。
它通過自洽場(chǎng)(SCF)計(jì)算電子波函數(shù),是更高精度方法的基礎(chǔ)。
適合:
小體系的幾何結(jié)構(gòu)優(yōu)化;
作為MP2或CCSD計(jì)算的前導(dǎo)步驟。
不足之處在于未考慮電子相關(guān),能量偏高。
?建議:用于結(jié)構(gòu)優(yōu)化或提供參考波函數(shù)。
3. 密度泛函理論(DFT)方法
DFT以電子密度為核心變量,在效率與精度之間取得良好平衡,是目前最常用的計(jì)算方法。
常見泛函及特點(diǎn):
B3LYP:經(jīng)典穩(wěn)健,適用于大多數(shù)有機(jī)分子體系;
M06系列:對(duì)金屬、π–π作用、非共價(jià)相互作用有較好表現(xiàn);
wB97XD:考慮長(zhǎng)程修正與色散作用,適合大體系或弱相互作用研究。
?建議:B3LYP作為默認(rèn)起點(diǎn),若體系含金屬或弱相互作用明顯,可選M06或wB97XD。
4. MPn微擾理論(MP2、MP3、MP4…)
在HF基礎(chǔ)上逐步引入電子相關(guān)修正:
MP2:計(jì)算精度高且成本適中,是最常用的相關(guān)修正方法;
MP3/MP4:理論上更精確,但計(jì)算量急劇增加,實(shí)際應(yīng)用較少。
?建議:MP2適用于中小體系的高精度能量計(jì)算。
5. 耦合簇(Coupled Cluster, CC)方法
被譽(yù)為“量子化學(xué)的金標(biāo)準(zhǔn)”,計(jì)算精度極高。
CCD:僅考慮雙激發(fā);
CCSD:加入單激發(fā)修正;
CCSD(T):進(jìn)一步微擾三激發(fā),是目前最精確的主流方法之一。
計(jì)算量非常大,通常僅用于小分子的基準(zhǔn)計(jì)算。
?建議:當(dāng)需與實(shí)驗(yàn)嚴(yán)密對(duì)比或驗(yàn)證其它方法可靠性時(shí)使用CCSD(T)。
二、基組的選擇
最小基組(Minimal Basis Set)
代表:STO-3G
每個(gè)原子軌道只用一個(gè)高斯函數(shù)近似;
計(jì)算量極低,但精度有限。
?用途:快速測(cè)試或教學(xué)演示,不推薦科研正式計(jì)算。
2. 分裂價(jià)基組(Split-Valence Basis Set)
代表:3-21G、6-31G、6-311G
將價(jià)電子軌道拆分為多個(gè)函數(shù),以更好描述電子分布;
是結(jié)構(gòu)優(yōu)化與能量計(jì)算的常用起點(diǎn)。
?建議:6-31G或6-31G(d) 是入門常用基組,可平衡精度與速度。
3. 極化函數(shù)與彌散函數(shù)
極化函數(shù)(d, p, f):用于更精確描述化學(xué)鍵方向性與分子畸變(如彎曲鍵、過渡態(tài));
彌散函數(shù)(+或++):用于描述電子云分布較寬的體系(如陰離子、氫鍵、π–π作用)。
?示例:
6-31G(d,p):常規(guī)優(yōu)化首選;
6-31+G(d,p):適合含陰離子或弱相互作用體系。
4. 相關(guān)一致基組(Correlation-Consistent Basis Set)
代表:cc-pVDZ、cc-pVTZ、cc-pVQZ
由Dunning提出,能系統(tǒng)改善電子相關(guān)描述;
常用于高精度計(jì)算(MP2、CCSD等)。
?建議:MP2或CCSD計(jì)算中使用cc-pVTZ可獲得較可靠能量。
5. 有效芯勢(shì)(ECP)基組
代表:LANL2DZ、SDD、Def2系列
通過勢(shì)函數(shù)近似內(nèi)層電子效應(yīng),顯著降低含金屬體系的計(jì)算量。
?建議:
含過渡金屬體系 → 使用LANL2DZ或Def2-TZVP;
想兼顧精度與速度 → Def2-SVP是不錯(cuò)選擇。
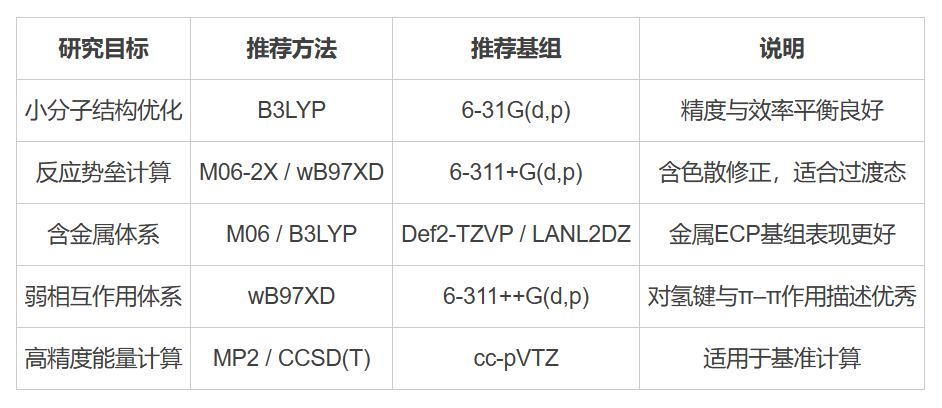
三、常見方法與基組搭配建議




 投訴建議
投訴建議
提交


分子動(dòng)力學(xué)的組成部分和用途有哪些?
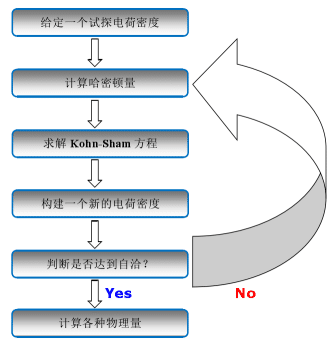
量子化學(xué)中密度泛函理論:從概念到應(yīng)用
量子化學(xué):什么是福井函數(shù)