


量子化學:HOMO/LUMO定義到應用
一、HOMO 與 LUMO 是什么?
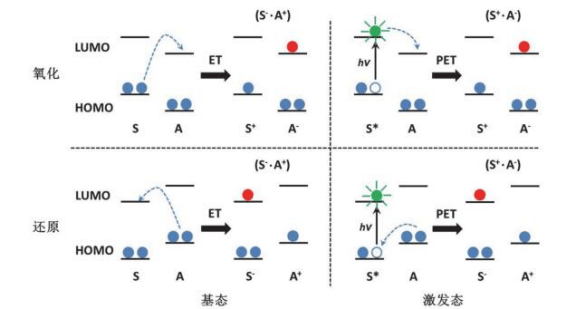
HOMO(Highest Occupied Molecular Orbital):最高已占據分子軌道,常對應最易被氧化或易失電子的能級。
LUMO(Lowest Unoccupied Molecular Orbital):最低未占據分子軌道,常對應最易被還原或易接受電子的能級。
兩者統稱“前線軌道(Frontier Orbitals)”。它們的能量差(能隙 ΔE)決定了分子被激發或發生反應的“難易度”。
二、為什么重要?三個高頻應用
(1) 、反應性預測:反應通道往往由反應物的 HOMO 與另一方的 LUMO 主導;能量接近、對稱性匹配且空間重疊良好時更有利于反應的發生,同時可以結合Fukui 函數、局部軟硬度 做位點選擇性預測。
(2) 、光譜與發光:最簡躍遷可近似為 HOMO→LUMO,能隙對應吸收/發射波長的粗略位置。
(3) 、電化學與能級:氧化電位常與 ε(HOMO) 相關,還原電位與 ε(LUMO) 相關;在固體中,類比為價帶頂與導帶底。
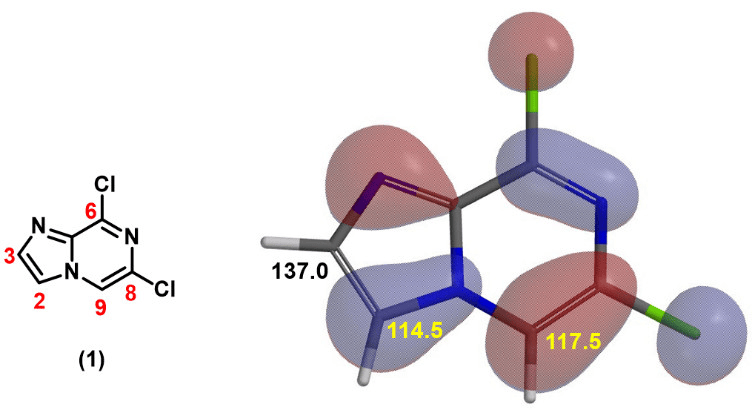
三、一個直觀例子:共軛 π 系統的 HOMO→LUMO 激發

隨著共軛長度增加,前線能級間距總體縮小,吸收/發射發生長波移。
取代基的給受電子效應會抬高或降低 HOMO/LUMO,從而調控顏色與反應活性。
在有機發光材料與染料設計中,適度的 HOMO/LUMO空間分離可降低交換能、調控三重態收發率。
四、如何“看見”與“估算” HOMO/LUMO?
實驗側:
- 循環伏安(CV):將氧化/還原起始電位換算到真空能標,近似估 ε(HOMO)和ε(LUMO),須注明參比電極、溶劑/電解質與溫度并做校正。
- UV-Vis/PL:吸收邊與發射峰反映光學隙與斯托克斯位移;先由 CV 定位 HOMO,再用帶隙估 LUMO。
計算側:
- 量化化學(DFT/HF/半經驗):輸出軌道能量與等值面,Koopmans 近似在 HF 更成立,DFT 僅作定性。
- 開殼層體系注意 SOMO/自旋分辨;溶劑、色散校正、構型與扭轉角都會顯著影響前線能級與隙,應在相同條件下對比。




 投訴建議
投訴建議
提交

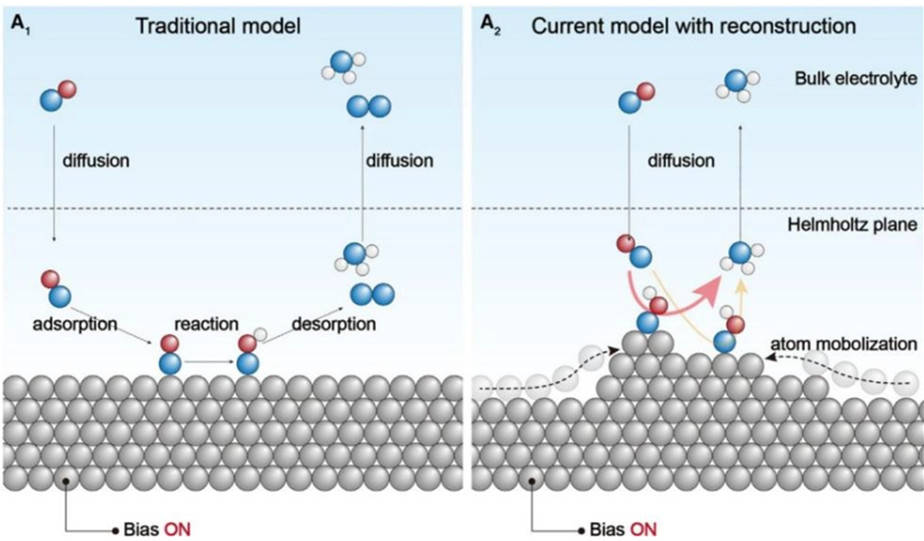
量子化學:什么是表面重構?
Gaussian溶劑模型,顯式與隱式

COMSOL仿真,通過二維平面創建三維體
金屬、半導體、絕緣體的區別與VASP模擬
第一性原理:磁性材料的分類及計算方法